|
DISTROFIAS CORNEALES HEREDITARIAS
Se subdividen a su vez en:
- Distrofias Corneales Hereditarias Superficiales
- Distrofias Corneales Hereditarias Estromales
- Distrofias Corneales Hereditarias Endoteliales
a) Distrofias Corneales Hereditarias Superficiales: del epitelio y membrana de Bowman
|
|
Distrofia de Meesman
Se transmite por herencia autosómica dominante. Es un trastorno de evolución lenta caracterizado por la existencia de quistes puntiformes intraepiteliales situados por delante de la membrana de Bowman.
Produce una serie de opacidades dispersas que no afectan a la visión de manera importante y se acompaña de modificaciones de la membrana basal; la membrana de Bowman está intacta. Se inicia en los primeros años de la infancia y cursa con episodios agudos de irritación ocular por erosiones recidivantes del epitelio.
|
|
|
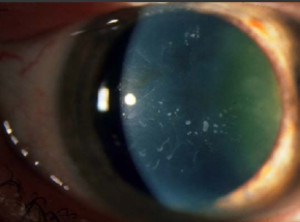
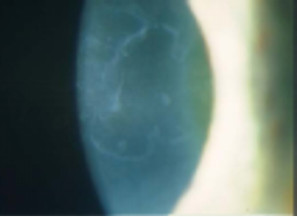
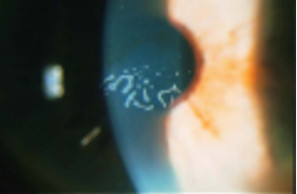
Distrofia de Cogan (Distrofía epitelial microquística)
Se transmite a veces por herencia dominante y es más común en las mujeres. Aparece en la adolescencia y se caracteriza por erosiones recidivantes. Es bilateral y se forman unas opacidades discretas intraepiteliales en la región pupilar que adoptan la forma de puntos o de líneas de huellas digitales grisáceas.
Estas opacidades corresponden a quiste que se depositan en un epitelio con células alteradas, edematosas. La membrana basal a menudo está engrosada y deformada, per la membrana de Bowman siempre es normal.
|
|
|
Erosión corneal atraumática recidivante
Aparece en la infancia y se caracteriza por la presencia de erosiones recidivantes que se acompañan de fotofobia intensa, pérdidas de substancias epiteliales (que se asocian a la existencia de quistes epiteliales) y ampollas intraepiteliales.
Desde el punto de vista histológico coexisten dos tipos de lesiones: quistes y defectos de reconstrucción de la membrana basal y de los hemidesmosomas.
|

|
|
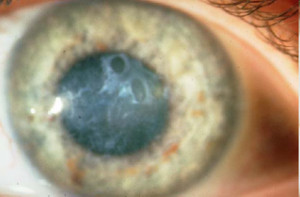
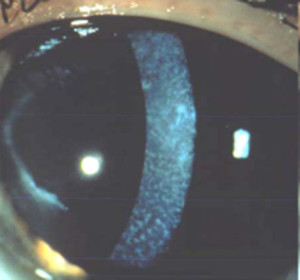
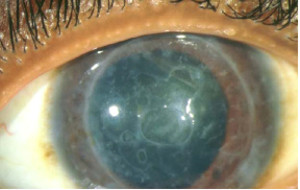
Distrofia de Reis-Bücklers (Reis-Büclers tipo I)
Se transmite por herencia autosómica dominante. Es un trastorno que afecta de forma importante a la membrana de Bowman, la cual al final es reemplazada por tejido conectivo celular que contiene colágena y material fibrilar o granular.
La córnea presenta opacidades subepiteliales grises en panal de miel a nivel de la membrana de Bowman que tienden a ser más densas en la región central. El epitelio suprayacente es irregular y de espesor variable y generalmente no se observa vascularización.
Es una afectación bilateral y simétrica y se inicia durante la primera década de la vida con síntomas de erosión recurrente. Progresa lentamente y conduce a una disminución de la agudeza visual, que puede ser marcada.
El material fibrilar depositado puede extirparse a través de una disección roma, procedimiento que se repite si el trastorno recidiva.
|
|
|
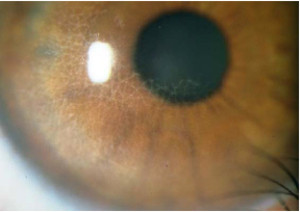
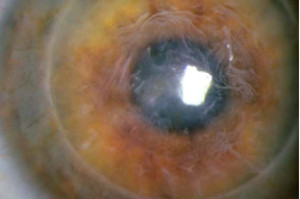
Distrofia en Panal de Thiel y Behnke (Reis- Büclers tipo II)
Subepitelial y bilateral, se transmite de forma autosómica dominante. Coexiste con un patrón patológico propio de la enfermedad de Reis-Bücler.
Comienza en la infancia con úlceras corneales que cesan expontáneamente entre la 40 y 60 decada de la vida, al evolucionar altera la membrana de Bowman produciendo una imágen en panal.
|
|
|
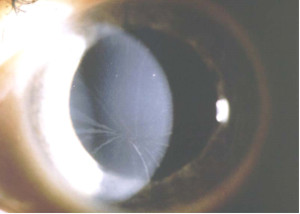
Distrofia de Fleisher-Grüber
También se llama distrofia verticilata y es característica de los portadores de la enfermedad de Fabry, en los que se transmite por herencia ligada al sexo. Se caracteriza por la presencia de líneas pigmentadas radiales,
formadas por glicolípidos, a nivel de la membrana de Bowman y estroma
anterior que se diseminan sobre toda la superficie de la córnea.
Suele ser bilateral y la agudeza visual no está muy afectada. Aparece en individuos jóvenes y sin signos funcionales. Aunque es típica de la enfermedad de Fabry, también puede producirse por depósitos de sustancias medicamentosas (fenotiacinas, cloroquina, indometacina, amiodarona, petidina, ...).
|
|
b) Distrofias Hereditarias Estromales
|
|
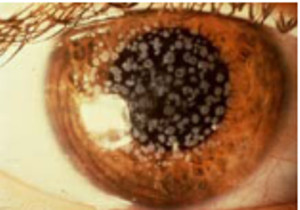
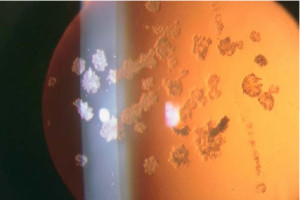
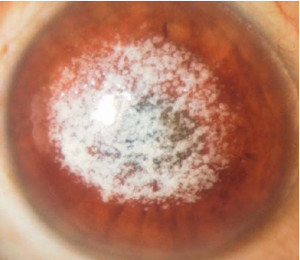
Distrofia granular (Groenow tipo I)
Se transmite por herencia autosómica dominante.
Se caracteriza por la aparición de lesiones blanquecinas, finas y granulares en la zona central del estroma anterior de la córnea, progresando hacia la periferia y las capas más posteriores.
Entre estas lesiones hay zonas de córnea intactas. Histológicamente puede verse un depósito uniforme de material hialino.
Aparece de forma precoz (es frecuente su inicio durante la infancia) y suele ser asintomática y de evolución lenta, de manera que solamente afecta a al agudeza visual de forma tardía.
|
|
|
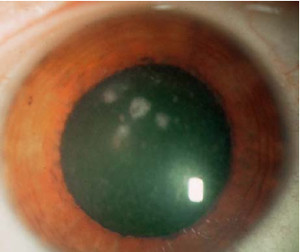
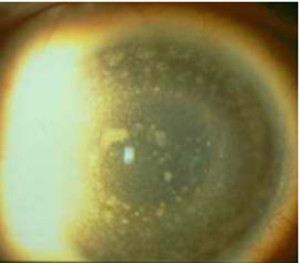
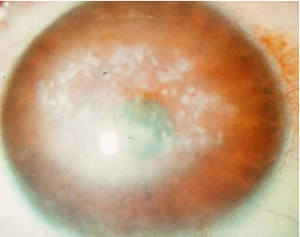
Distrofia Macular (Groenow tipo II)
Se transmite por herencia autosómica recesiva.
Se caracteriza por la presencia de depósitos blanquecinos mal delimitados que confluyen entre si, de aspecto turbio y que ocupan todo el espesor
del estroma corneal, respetando la zona prelímbica. A nivel histológico puede verse un depósito de mucopolisacáridos ácidos en el estroma y degeneración de la membrana de Bowman.
Es una degeneración mucho más rara que las anteriores y es bilateral y simétrica. Aparece en la primera década de la vida y comporta una disminución profunda y precoz de la agudeza visual y erosiones recurrentes de la córnea.
Es frecuente la existencia de una córnea guttata asociada.
|
|
|
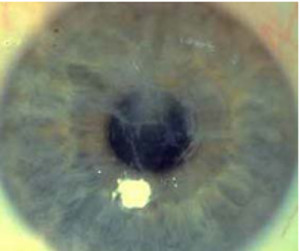
Tipo I (Biber-Haab- Dimmer)
La herencia es autosómica dominante. Es una forma localizada de amiloidosis con inicio temprano en la vida (20-30 años de edad), que afecta a ambos sexos. Se manifiesta por la progresiva opacidad de la córnea formando una red de celosía (al igual que filamentos de ramificados).
Las opacidades pueden esparcirse por toda la córnea, pero las zona interlesionales son claras, y puede haber depósitos extracelulares de material amiloide. La función visual por lo general sigue siendo bastante satisfactorio durante mucho tiempo.
|

|
|
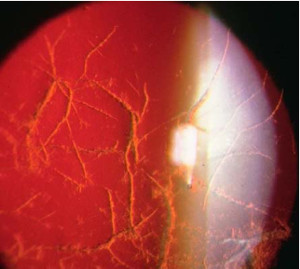
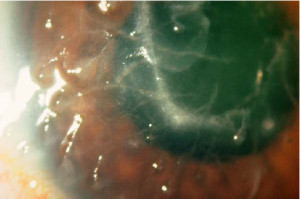
Típo II (Amiloidosis familiar o Síndrome Meretoja)
Se transmite por herencia autosómica dominante.
Se caracteriza por la existencia de unas estriaciones lineales de coloración amarillenta que se entrecruzan en forma de enrejado. Están localizadas en la zona central y afectan a las capas anteriores del estroma diseminándose hacia la periferia. El examen histológico revela depósitos amiloides en las fibras de colágeno. A veces, la acumulación de estos depósitos forma una condensación densa y amarillenta en la zona central.
|
|
|
Distrofia Reticular
La distrofia reticular es una enfermedad de herencia autosómica dominante, bilateral, simétrica y lentamente progresiva. Su
sintomatología se inicia generalmente en la primera década de la vida en forma de erosiones recurrentes o dificultad en la visión.
En estadíos iniciales pueden apreciarse puntos y líneas finas irregulares y depósitos en estroma anterior y subepiteliales. Más tarde estos depósitos lineales traslúcidos de sustancia amiloide (líneas de Lattice) se alargan y se entrelazan a nivel del estroma, dándole a la córnea un aspecto de vidrio esmerilado. El estroma entre ellas es claro al principio, pero progresivamente se vuelve más opaco. En esta fase, las erosiones recidivantes dejan de producirse, aunque la agudeza visual disminuye. La sensibilidad corneal está disminuida. Los depósitos son amiloides, se tiñen con rojo Congo y presentan birrefringencia verde con microscopio de luz polarizada.
|


|
|
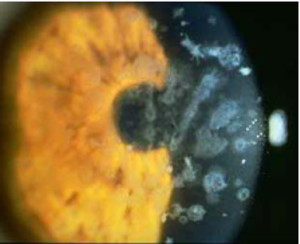
Distrofia Combinada Granular-Reticular (Avellino)
En este tipo de distrofia se mezclan la distrofia granular y la reticular en la misma córnea y se puede acompañar de queratocono, el patrón de
transmisión es autosómico recesivo con penetrancia variable y su aparición es durante la primera década de la vida. Apareció en una zona de Italia conocida como Avellino.
Recientes descubrimientos genéticos y de biología molecular han agrupado distrofias corneales como la de Reis-Bückler, Thiel-Benke, granular, reticular y de Avellino en un mismo grupo etiopatogénico, al presentar la misma mutación del gen BIGH3 ubicado en el brazo largo del cromosoma 5 que condiciona depósitos de queratoepitelina, una proteína cuyo origen parece epitelial aunque acaba acumulándose en el estroma corneal. Las frecuentes recidivas a largo plazo de estas distrofias tras queratoplastia penetrante pueden actualmente atribuirse a la sustitución que sufre el epitelio donante por parte del receptor en todos los casos.
|
|
|
Distrofia Combinada Granular-Reticular (Hida)
Es otro tipo de combinación de distrofia granular y reticular. Aquí los trazos reticulares son mas acusados y groseros que en la de Avellino.
|

|
|
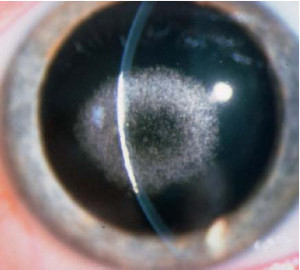
Distrofia Cristalina Central (Schnyder)
Se presenta de manera autosómica dominante, existe acumulo de cristales en el centro corneal en forma bilateral que aparece en el primer año de vida. Tiene dos asociaciones sistémicas frecuentes, genu valgum e hiperlipidemia. La densidad de la opacidad corneal no se correlaciona con el nivel de lípidos en suero. Permanecen asintomáticos hasta que se forma una densa opacidad corneal central entre la tercera y la cuarta décadas de la
vida. Sin embargo, la apariencia clínica varía ampliamente. La sensibilidad corneal puede estar disminuida
Esta distrofia tiene cuatro componentes clínicos que se presentan en diferentes combinaciones: cristales minúsculos, opacidad gris estromal difusa, un arco corneal denso y un anillo limbar, aproximadamente el 50% de todos los pacientes presentan la variedad de cristales minúsculos. Las opacidades centrales se acumulan por detrás de la Bowman, algunas veces extendiéndose en el estroma profundo y ocasionalmente se fusionan con el arco corneal.
El patrón en disco consiste en una opacidad amorfa sin cristales, o de cristales en la periferia, o bien de cristales orientados en forma radial junto a un anillo de opacidad en la periferia. El patrón en anillo consiste de material gris amorfo ausente de cristales o sólo con algunos presentes.
Los cristales finos semejan fibra de vidrio en diferentes colores. Se agregan en forma de esponja, o crean una palizada periférica. El estroma que no se ha afectado permanece claro, aunque en ocasiones existen pequeñas opacidades blanquecinas puntiformes.
|

|
|
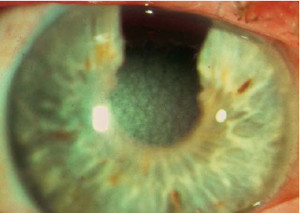
Distrofia Puntíforme (Françoís-Neetens)
Es una distrofia conocida también como en moteado, es de tipo autosómico dominante, y ha sido detectada desde el segundo año de vida, aunque
puede ser congénita y progresa lentamente. Es generalmente bilateral, sin embargo se puede encontrar gran asimetría o incluso unilateralidad.
Debido a lo pequeño de las opacidades y a que su superficie es lisa, éstas no disminuyen la agudeza visual y por lo general el oftalmólogo es quien descubre la distrofia de manera incidental.
Se ha encontrado en algunos individuos, la asociación de opacidades corticales puntiformes en el cristalino que acompañan a la distrofia. En otros casos se ha encontrado la distrofia puntiforme acompañada con la distrofia nebulosa central, queratocono, dermoide limbal, disminución en sensibilidad corneal y pseudoxantoma elástico. Estas opacidades puntiformes blanco grisáceas se extienden en todo el estroma corneal y escasamente en capa de Bowman algunas llegan a alcanzar el limbo. Excepcionalmente, las opacidades ocupan el centro o la periferia estromal. El número de opacidades varía de escasas a difusas en la totalidad de la córnea. La forma de las opacidades es más aparente en iluminación focal directa en alta magnificación. Con frecuencia la opacidad blanquecina clara se ve rodeada de un anillo blanco. La arquitectura estromal y los queratocitos permanecen normales. Es posible encontrar escasos queratocitos aislados que contienen dos sustancias anormales, exceso de glucosaminoglicanos que tiñen azul con Azul Alciano y Hierro Coloidal y lípidos que se demuestran con Negro Sudán y con Aceite Rojo 0. El epitelio, capa de Bowman, membrana de Descemet y endotelio son normales. La base metabólica para esta anormalidad selectiva es desconocida. Por el momento ningún tratamiento es requerido para la distrofia puntiforme
|
|
|
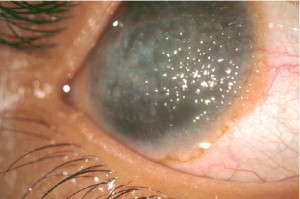
Distrofia Gelatinosa en Forma de Gotas
Es una forma de amiloidosis familiar primaria de la córnea, se ha observado en Estados Unidos y Europa pero la frecuencia es mayor en Japón, aparentemente tiene herencia autosómica recesiva. La distrofia aparece en la primera década de vida se presenta con fotofobia lagrimeo y disminución de la agudeza visual. Aparecen montículos subepiteliales de amiloide bilaterales, centrales, elevados y multinodulares. Aparecen blancos con la iluminación directa y transparentes a la retroiluminación.
En etapas tempranas, los depósitos están aplanados semejan una queratopatía en banda, en estados tardíos la córnea adquiere un aspecto difuso aframbuesado y puede contener neovasos. Histológicamente se ha demostrado que los depósitos corresponden a amiloide, suele no existir capa de Bowman y el amiloide se deposita en las células epiteliales básales, debajo del epitelio y en estroma. La queratoplastia penetrante no es muy eficaz pues recidiva.
|
|
|
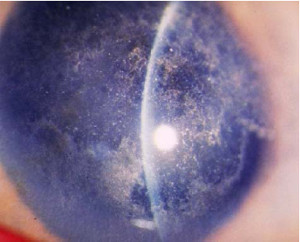
Distrofia Cristalina (Bietti)
Fue descrita en 1937 como depósitos marginales de cristales en el estroma periférico Anterior y en la retina paracentral y peripapilar. Cambios pigmentarios se observan en la fóvea y en la retina periférica, el patrón de transmisión no ha sido descrito, sin embargo su mayor incidencia en hijos de matrimonios consanguíneos sugieren herencia autosómica recesiva.
El origen de los cristales es desconocido, algunos cristales semejan colesterol, ésteres de colesterol u otros lípidos en fibroblastos corneales, conjuntivales y en linfocitos por lo que sugiere una anormaIidad sistémica en el metabolismo de los lípidos. No se observa disminución de la agudeza visual, por lo que no se requiere tratamiento, salvo casos muy avanzados.
|
|
Distrofias Hereditarias Endoteliales
|
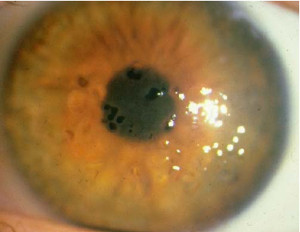
Distrofia Endotelial Hereditaria Tardía (Fuchs)
Herencia autosómica dominante que produce una condición degenerativa del endotelio corneal.
La afectación es inicialmente endotelial, y termina alterando a la totalidad de las capas, con enturbiamiento corneal por el edema y que a menudo se complica con glaucoma de ángulo abierto. La incidencia es mayor en mujeres a partir de la 5ª o 6ª década de la vida. Es bilateral y progresiva, y cursa con córnea guttata, edema corneal y queratopatía bullosa.
Histopatológicamente puede apreciarse una escasez de células endoteliales. El endotelio corneal puede evaluarse mediante la microscopía especular. Esta monocapa endotelial no se reproduce in vivo y va disminuyendo con la edad desde unas 3500 células/mm2 en el neonato hasta unas 2000 en el adulto. La descompensación corneal se hace más probable con una densidad inferior de 500 células/mm2
El fallo endotelial da lugar a los síntomas, que incluyen disminución de visión, sensación de cuerpo extraño y dolor.
Hallazgos biomicroscópicos:
Cornea Guttata: son excrecencias de la membrana de Descemet debidas al depósito excesivo de colágeno y que se hacen más frecuentes con la edad.
Edema corneal estromal: producido por el empobrecimiento endotelial. Si el edema estromal supera el 30%, se producirá además un edema epitelial con la formación de bullas y microquistes. Estos, al romperse, producen dolor y sensación de cuerpo extraño, acompañado de lagrimeo y fotofobia.
|



|
|
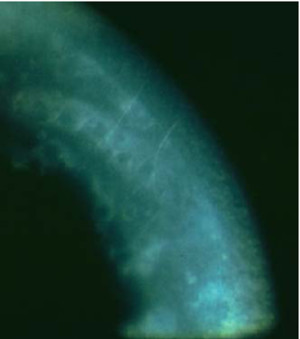
Distrofia Polimorfa Posterior
Es una distrofia endotelial de carácter familiar con herencia autosómica dominante. Clínicamente se aprecian unas opacidades localizadas en la cara posterior de la córnea, bilaterales y con cierta simetría en su morfología. Su desarrollo es progresivo a lo largo de la vida.
Morfológicamente se caracteriza por la existencia de lesiones nodulares, vesiculares y/o ampollosas de 2 a 20; o también opacidades planas de color gris-blanco. La cara anterior de la Descemet es de morfología normal, mientras que la cara posterior se encuentra engrosada, con zonas de aumento de la matiz extracelular de colágeno y otras con ausencia de esta.
Por biomicroscopia especular podemos advertir bandas endoteliales oscuras, amplias, sinuosas con bordes irregulares (snail tracks), de morfología geográfica, con márgenes en ocasiones festoneados. Estas lesiones tienen un aspecto característico por retroiluminación, vesicular o en piel de naranja o como metal batido. Podemos encontramos también áreas redondeadas oscuras, con alguna célula en medio dando apariencia de «donut», que algunos autores indican como prácticamente patognomónico de la polimorfa posterior.
|

|
|
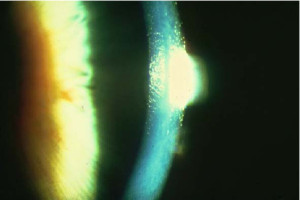
Distrofia Endotelial Congénita Hereditaria
Es una distrofia con herencia autosómica dominante o autosómica recesiva, se han reportado casos aislados. Los pacientes con patrón autosómico recesivo manifiestan la enfermedad desde el nacimiento, permanece estacionaria, asintomática y se acompaña de nistagmo. En los casos autosómicos dominantes se manifiestan en el primero a segundo año de vida con fotofobia y lagrimeo, progresa lentamente en los primeros diez años y sin presentar nistagmo. Presenta edema estromal y epitelial con engrosamiento grisáceo a nivel de la capa de Descemet, da un aspecto "en piel de naranja".
En algunos casos se alcanza a percibir el mosaico endotelial, sin embargo, se pueden apreciar espacios ausentes endotelio. En el período neonatal, el médico puede confundirse con el cuadro y sospechar un glaucoma congénito sin buftalmos. Sin embargo, la ausencia de inflamación, fotofobia y crecimiento corneal, así como la ausencia de presión intraocular elevada, lo descarta. En general, el espesor corneal en una distrofia corneal congénita hereditaria es de 2 a 3 veces mayor de lo normal. En ocasiones se pueden apreciar manchas grisáceas focales prominentes, que pueden simular una distrofia macular, aunque la edad de presentación las diferencia. El manejo de estos pacientes en un inicio puede ser usando soluciones hipertónicas,
afortunadamente las erosiones epiteliales no son dolorosas. Sin embargo, debido al edema corneal tan importante y a la opacificación, se sugiere queratoplastía penetrante. Las recurrencias en el injerto son altas por lo que el pronóstico visual de estos pacientes es sombrío
|


|
|

























